Pharmacological Chaperone Database
Proteome-scale computational database of de novo pharmacological chaperones for pathogenic protein misfolding variants, generated by the REFOLD pipeline.
Protein Misfolding & the Pharmacological Chaperone Gap
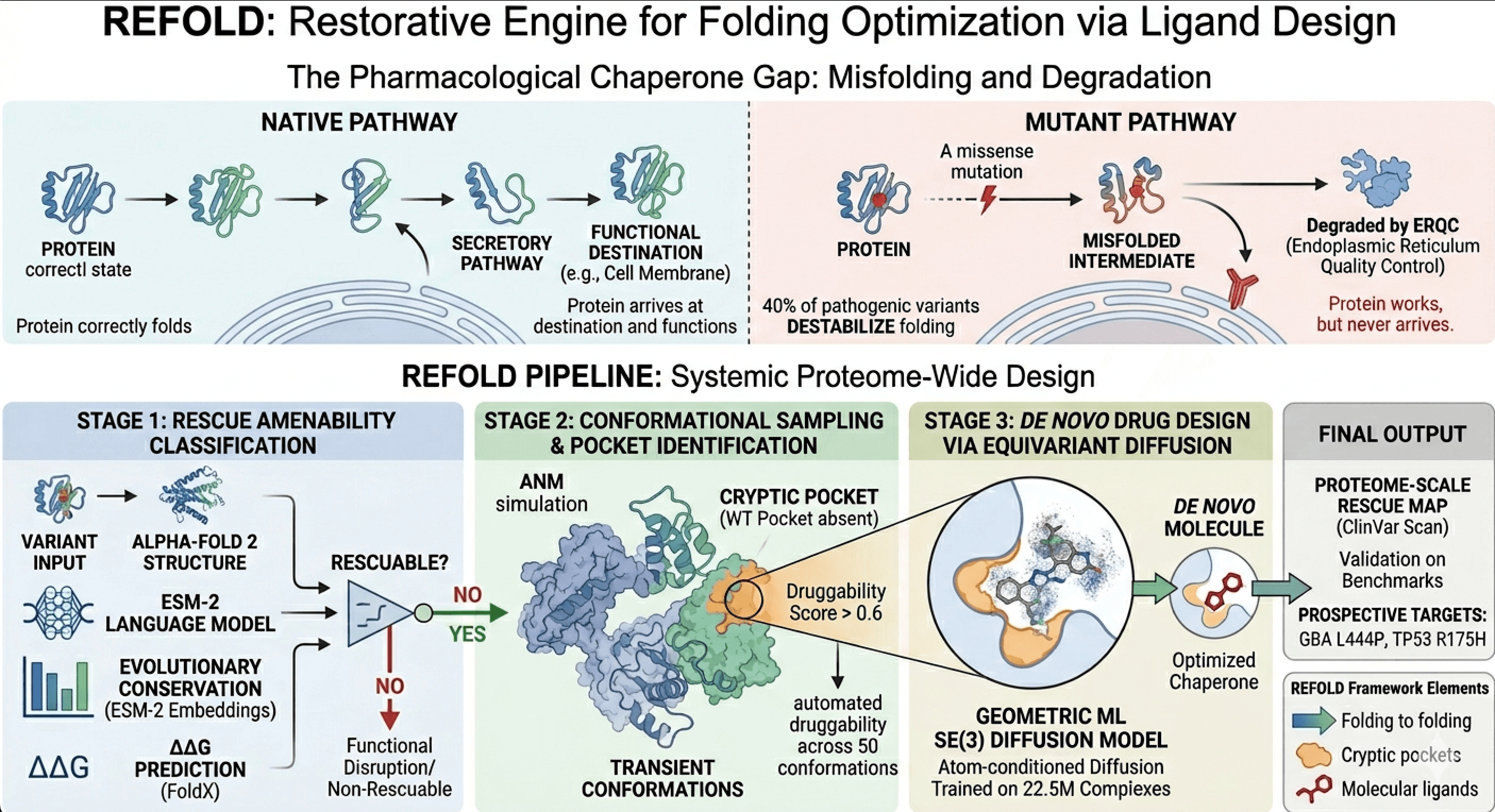
40–60% of pathogenic missense variants cause disease not by disrupting a protein's active site, but by triggering premature proteasomal degradation of the misfolded intermediate before it can reach its functional destination. The endoplasmic reticulum quality-control machinery flags structurally aberrant conformers and routes them for degradation via the ubiquitin-proteasome system.
The key mechanistic insight: these proteins retain native-like function; they simply never arrive. A small molecule that binds the misfolded intermediate and shifts the folding free-energy landscape toward the native basin constitutes a pharmacological chaperone. No systematic computational framework for their design has previously been described. REFOLD provides this framework.
Three FDA-approved pharmacological chaperones currently exist, each discovered by brute-force high-throughput screening against a single target at cost exceeding $1B per molecule. REFOLD designs them from structural first principles, for every protein in the human proteome, fully automatically.
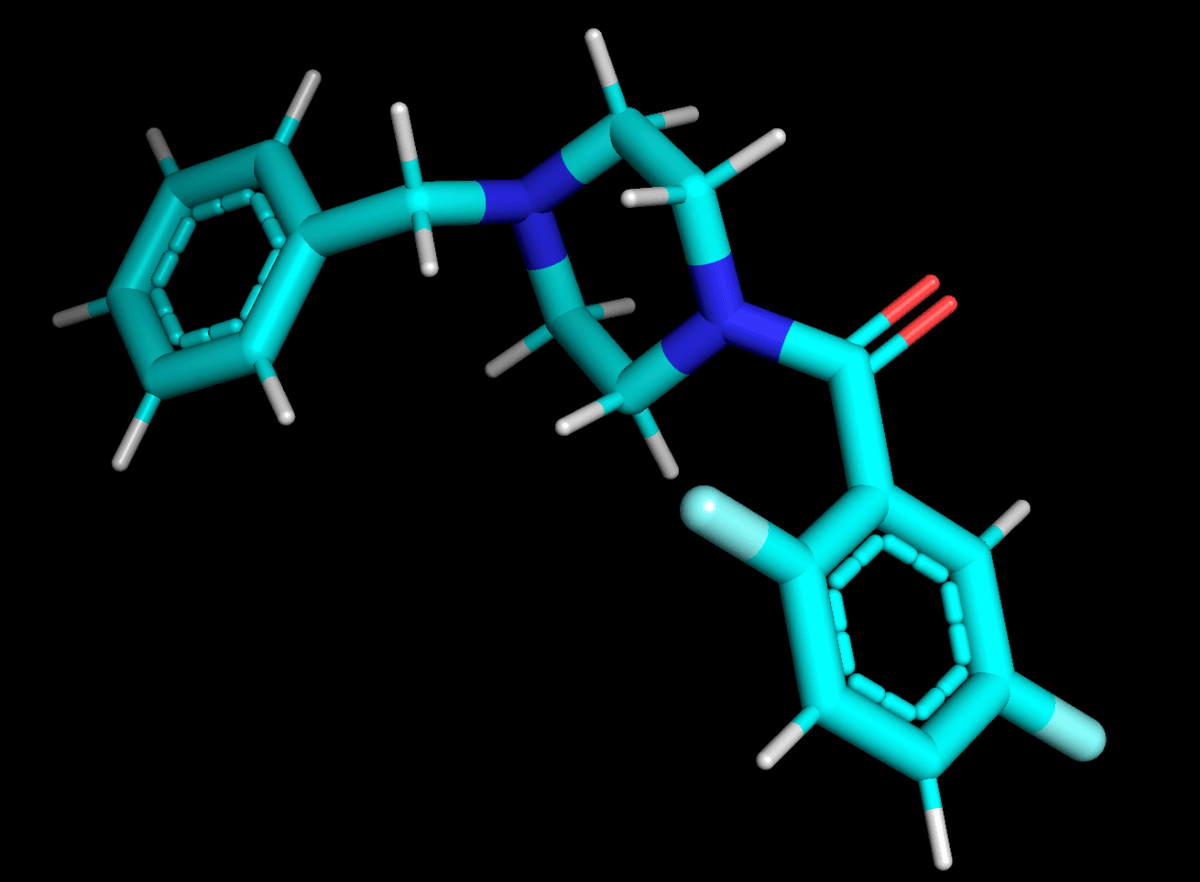
Representative transient pocket (cyan surface) identified in the misfolded intermediate of CFTR-F508del. De novo chaperone shown as amber sticks. Generated by REFOLD Stage 2–3.
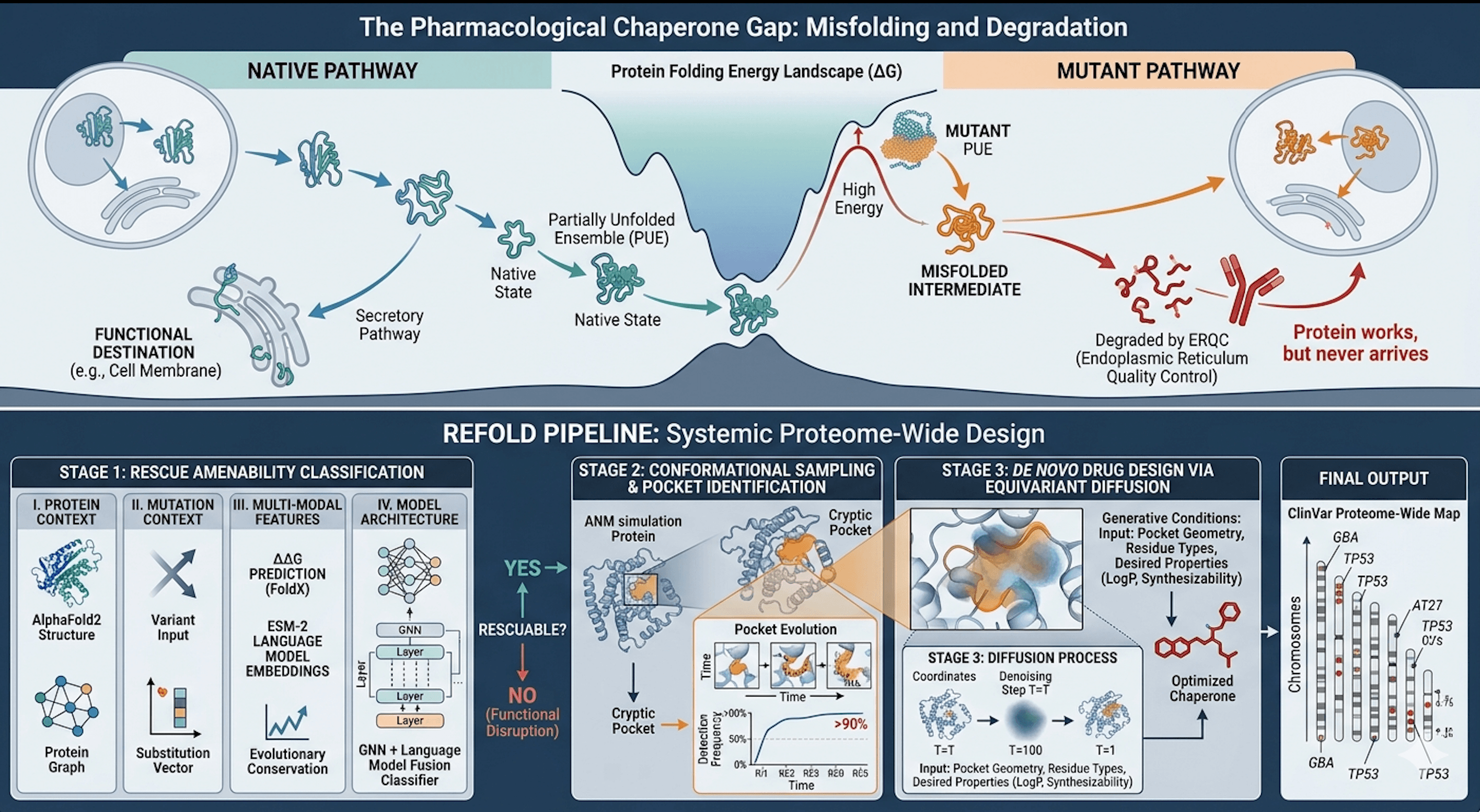
Top: Native vs. mutant folding free-energy landscape. Bottom: REFOLD Stage 1→2→3 pipeline producing the proteome-wide chaperone map.
Part A: GNN feature extractor with atom-edge message passing + Transformer LM fusion → rescuability logits.
Part B: SE(3)-equivariant denoising network for structure-conditioned molecular generation (T=100→T=1 diffusion trajectory).
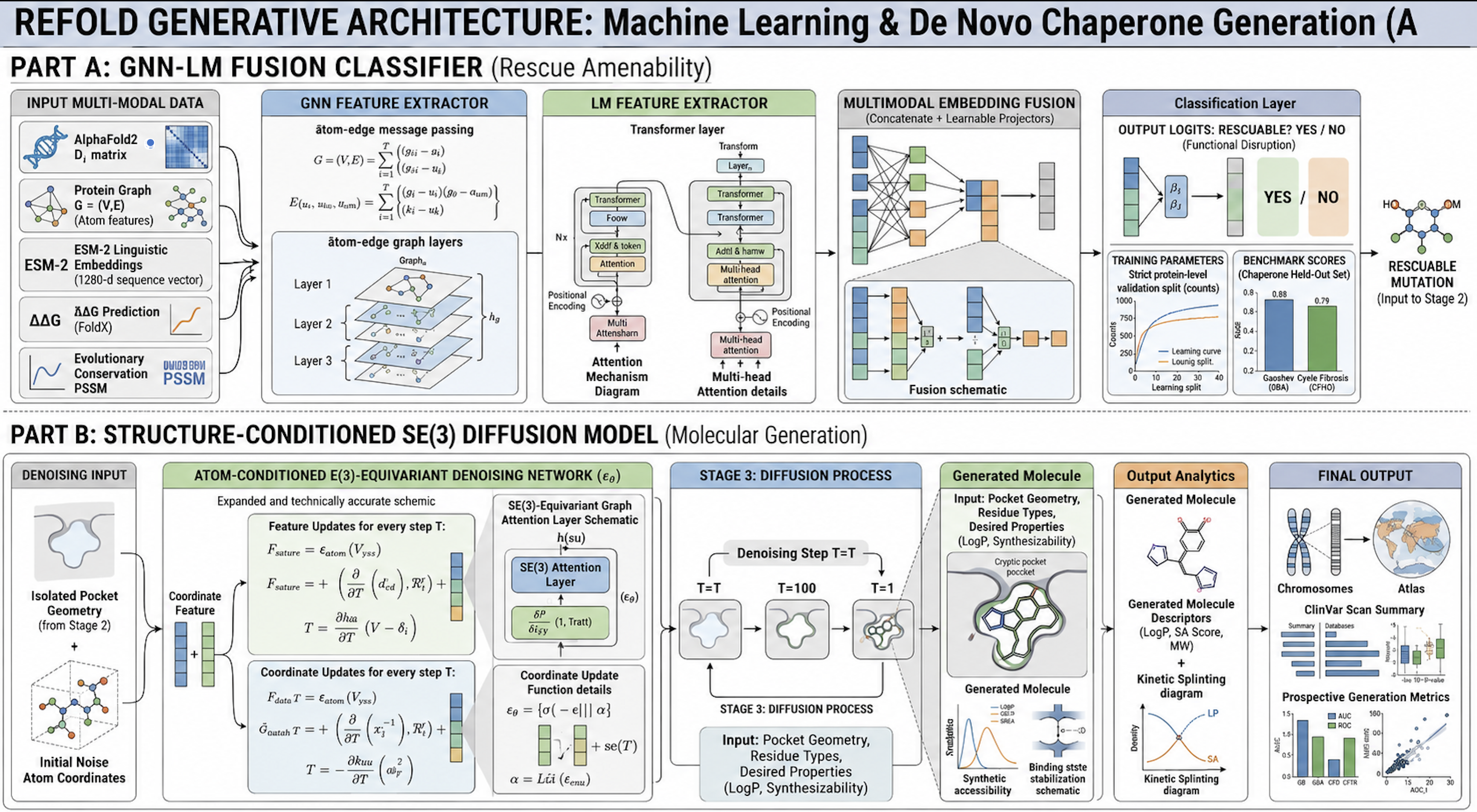
Inputs: AlphaFold2 structure, ESM-2 embeddings (1280-d), FoldX ΔΔG, PSSM conservation scores. Conditioning: E_ij Cα–Cα distance matrix from Stage 2 pocket geometry.
Chaperone Entries
6,316 entries · sorted by fpocket druggability score (desc.)

REFOLD — Proteome-Scale Pharmacological Chaperone Design
REFOLD is a fully automated, end-to-end computational pipeline that accepts any human disease-associated missense mutation as input and produces: (1) a binary prediction of whether the variant drives pathogenesis via protein misfolding rather than active-site disruption; (2) the 3D transient-conformation structure of the mutant protein with cryptic binding pockets resolved; (3) de novo designed small-molecule candidates that bind those pockets and thermodynamically stabilize the native fold.
The PCD is the product of continuously running REFOLD across the complete human proteome — processing every pathogenic missense variant in ClinVar sequentially, with each result automatically injected into this living database.